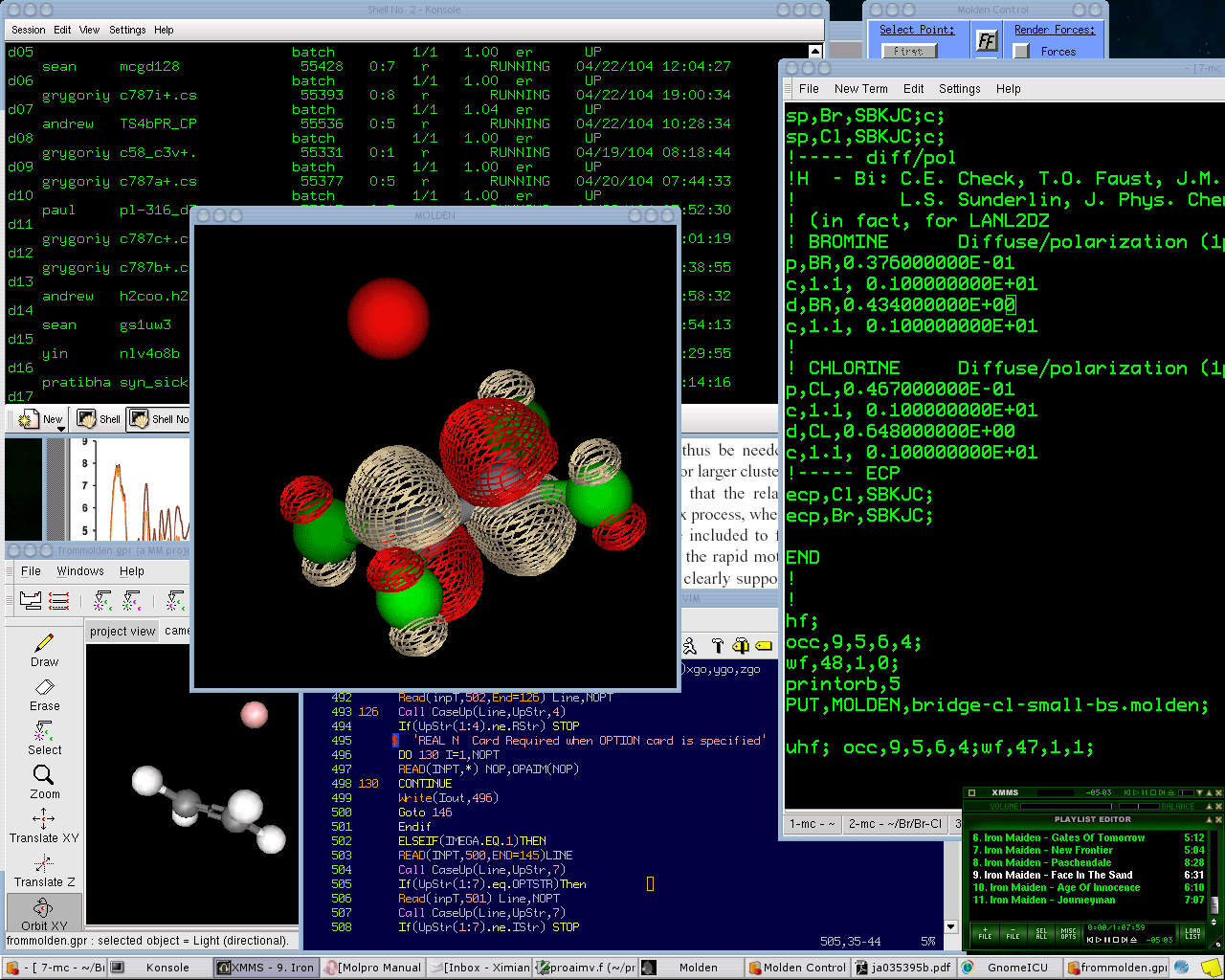
Квантовая химия традиционно делается на Unix-системах.
Мой домашний компьютер (один из): PIII/600 MHz, 512 MB, Vodoo3, Gentoo 2004.0, Kernel 2.6.3, XFce 4.0.4.
Konsole запущен _удаленно_ (по ADSL совершенно не тормозит) на одном из компьютеров используемых для входа на наш вычислительный кластер. Кластер - около 100 машин разного рода: двухпроцессорные третьи пни, однопроцессорные четвертые, двухпроцессорные Xeon'ы; памяти - от 1 до 2 GB, Red Hat 7.1 - 9.0. Все это управляется древней DQS 3.2 (давно надо бы OpenPBS поставить). Как раз видно часть статистики DQS - где, что и кем считается.
MOLDEN как-то уже пробегал на LOR`е, весьма удобная прога для просмотра результатов, хоть и интерфейс страшноват. Выполняется, к стати, тоже удаленно, на AlphaServer ES40.
Ghemical (слева внизу) - в принципе аналог виндового Chem3D, но пока еще сыроват.
До сих пор еще куча квантово-химических программ написано (и продолжет писаться) на Fortran77 . Пытаюсь разобраться в одном не очень новом коде (в gvim'е).
Многие квантово-химические проги очень навороченные и стоят больших денег, например, MOLPRO (input file для него редактируется в multi-gnome-terminal).
Ну, еще Adobe Acrobat Reader (на заднем плане) и xmms (в дань традиции LOR'а).
<disclaimer> Konsole запущен удаленно. Да, мне нравяться кислотно-зеленые терминалы :) Фортрановский код в gvim'е - не мой. На этом скрине все сваленно в кучу с чисто демонстрационными целями, обычно все разложено по workspace`ам. </disclaimer>

