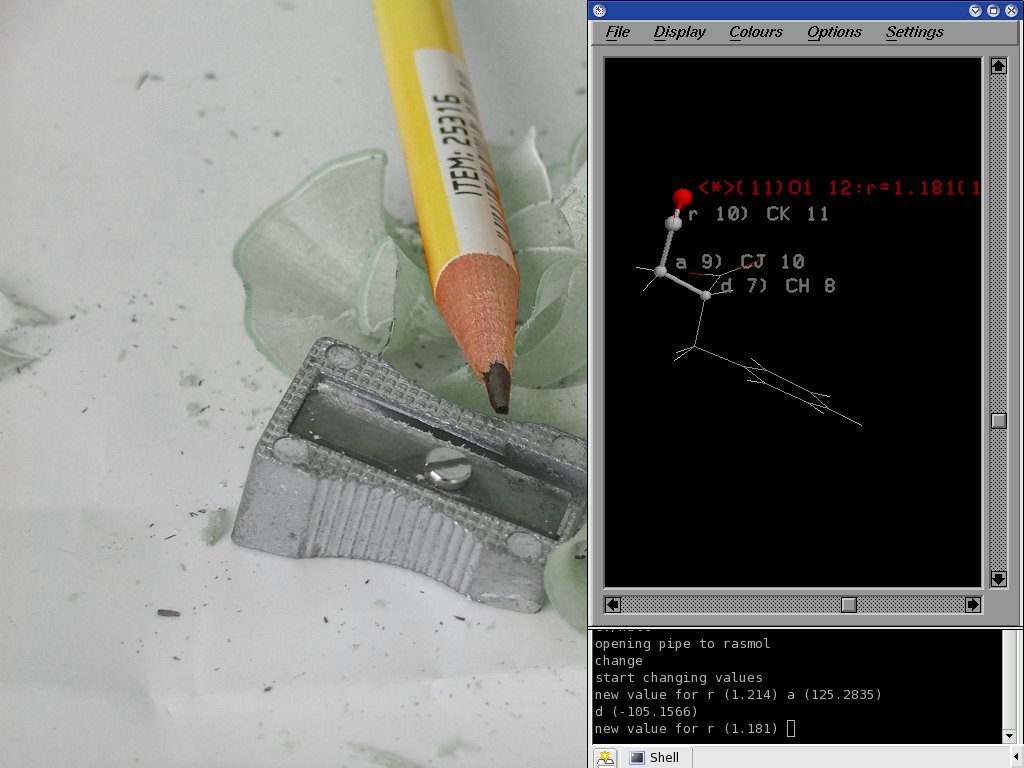
К своим программам, надо было изменять различные
параметры в иследуеммых молекулах. Автоматизировать
эту задачу не легко, так как каждый параметр зависит от очень
многих факторов. Вот мое решение проблеммы (все склеенно перлом):
1. берется файл с молекулой
2. вычесляется таблица связей (отдельная программа в С++)
3. вызывается rasmol, используя IPC::Open2. Нажатие на атом в rasmol-е
выдает сообщения на stdout, которые ловятся и обрабатываются. Перловый
скрипт помечает текущий атом и все нужные делу атомы и связи,
спрашивает как изменить параметры данного атома и готовит
файл параметров, который послужит вводом для действительно серьезных
програм.
Система: dual Xeon, mandrake 10.1 OE, КДЕ 3.2.3
Таскбар (а.к.а кикер) свернут в сторону
rasmol: http://www.rasmol.org/ (очень легкая и шустрая программа дла
визуализации молекул в формате PDB)
Молекулярное моделлирование - Unix way